Prion diseases or transmissible spongiform encephalopathies (TSEs) are a family of rare progressive neurodegenerative disorders that affect both humans and animals. They are distinguished by long incubation periods, characteristic spongiform changes associated with neuronal loss, and a failure to induce inflammatory response.
The causative agents of TSEs are believed to be prions. The term "prions" refers to abnormal, pathogenic agents that are transmissible and are able to induce abnormal folding of specific normal cellular proteins called prion proteins that are found most abundantly in the brain. The functions of these normal prion proteins are still not completely understood. The abnormal folding of the prion proteins leads to brain damage and the characteristic signs and symptoms of the disease. Prion diseases are usually rapidly progressive and always fatal.
Familial prion diseases of humans include classic Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), and fatal insomnia (FI). These conditions form a spectrum of diseases with overlapping signs and symptoms.
The prevalent controversial ‘protein-only’ hypothesis proposes that the infectious agent is a misfolded protein that triggers and propagates the disease in the absence of nucleic acid. The prion protein is the cause for all known mammalian prion diseases. Prion proteins are thought to exist in two different conformations. A properly folded form is denoted PrPC (for Common or Cellular) and a misfolded form is denoted PrPSc (for Scrapie, after one of the diseases first linked to prions and neurodegeneration.) The precise structure of the prion is not known but it can be formed by combining PrPC, polyadenylic acid, and lipids in a Protein Misfolding Cyclic Amplification (PMCA) reaction.
The protein encoded by the major prion protein gene is a membrane glycosylphosphatidylinositol-anchored glycoprotein that tends to aggregate into rod-like structures. The encoded protein contains a highly unstable region of five tandem octapeptide repeats. This gene is found on chromosome 20, approximately 20 kbp upstream of a gene which encodes a biochemically and structurally similar protein to the one encoded by this gene. Mutations in the repeat region as well as elsewhere in this gene have been associated with Creutzfeldt-Jakob disease, fatal familial insomnia, Gerstmann-Straussler disease, Huntington disease-like 1, and kuru. Alternative splicing results in multiple transcript variants encoding the same protein.
Neurodegenerative diseases such as Creutzfeldt-Jakob disease, Alzheimer’s disease (AD), Parkinson’s disease, and amyotrophic lateral sclerosis (ALS) share two remarkable characteristics. First: more than 80% of cases are sporadic. Second: many of the disease-specific mutant proteins are expressed in embryogenesis, however, the inherited forms of these neurodegenerative diseases are late-onset. Stanley Pruisner who received a Nobel price in Physiology or Medicine in 1997 for his research achievements in prion diseases offers an alternative unifying explanation shown in figure 1. He states that a diverse group of proteins can form prions. He explains that a small numbers of prions could be cleared by protein degradation pathways but accumulation of the prions above a certain threshold over time would enable the prions to self-propagate which will result in central nervous system (CNS) dysfunction.
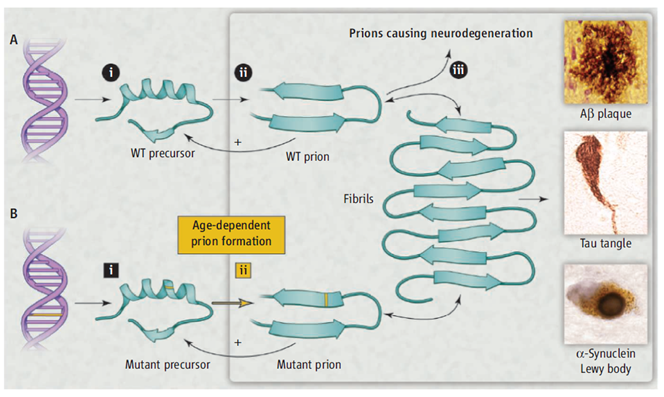
Figure 1: Modeling neurodegeneration caused by prions. (A) Wild-type (WT) prions multiply through self-propagating cycles of posttranslational modification; generally, an increase in ®-sheet content accompanies prion formation. Pathogenic prions are most toxic as oligomers and less toxic after polymerization into amyloid fibrils. Depending on the protein, the fibrils coalesce into amyloid plaques, neurofi brillary tangles, or intracellular inclusions such as Lewy or Pick bodies. Drug targets for the development of therapeutics (black circles): (i) lowering precursor protein, (ii) inhibiting prion formation, and (iii) enhancing prion clearance. (B) Late-onset heritable neurodegeneration argues for two discrete events (squares): (i) mutant protein synthesis and (ii) prion formation.
Adapted from: Stanley B. Prusiner; A Unifying Role for Prions in Neurodegenerative Diseases SCIENCE VOL 336 22 JUNE 2012 1511-1512.
The protein structures for several fragments of the whole prion protein have been solved. A few structure models are shown in Figure 2. Figure 3 and 4 show the sequence alignments for several prion proteins as well as the sequences for a few synthetic peptides used in prion research. Furthermore, the octarepeat containing the sequence PHGGGWGN present in the mature prion protein has been shown to bind to copper ions (Whittal et al 2000).
Figure 2: Structures of parts of the prion proteins as determined by either X-Ray crystallography or NMR. Source: NCBI protein structure database.
Figure 3: Graphical display of the prion superfamily proteins. Source: NCBI.
Figure 4: Sequence alignment of the prion superfamily proteins including synthetic peptides.
Legend: HPPP, ADD71057 prion protein, partial [Homo sapiens];
HMPPP, major prion protein preproprotein Homo sapiens NP_000302;
1QLX,
Chain A, Human Prion Protein; 3HES, Human Prion Protein Variant F198s With M129; 2KKG, Chain A, Octarepeat Region Of Prion Protein Bound To Pentosan Polysulfate; SPP, scrapie prion protein.
References:
- Aguzzi A, Heikenwalder M. Pathogenesis of prion diseases: current status and future outlook. Nat Rev Microbiol. 2006 Oct;4(10):765-75.
- Aguzzi A. Prion diseases of humans and farm animals: epidemiology, genetics, and pathogenesis. J Neurochem. 2006 Jun;97(6):1726-39.
- Belay E., Schonberger L., The Public Health Impact of Prion Diseases, Annu. Rev. Public Health 2005;26:191-212
- GRIFFITH, J. S.; Nature of the Scrapie Agent: Self-replication and Scrapie Nature 215, 1043 - 1044 (02 September 1967) NCBI
- Prusiner, Stanley B.; A Unifying Role for Prions in Neurodegenerative Diseases SCIENCE VOL 336 22 JUNE 2012 1511-1512
- Randy M. Whittal, Haydn L. Ball, Fred E. Cohen, Alam L. Burlinggame, Stanley B. Prusiner, and Michael A. Baldwin, Copper binding to octarepeat peptides of the prion protein monitored by mass spectrometryProtein Science (2000), 9:332–343.